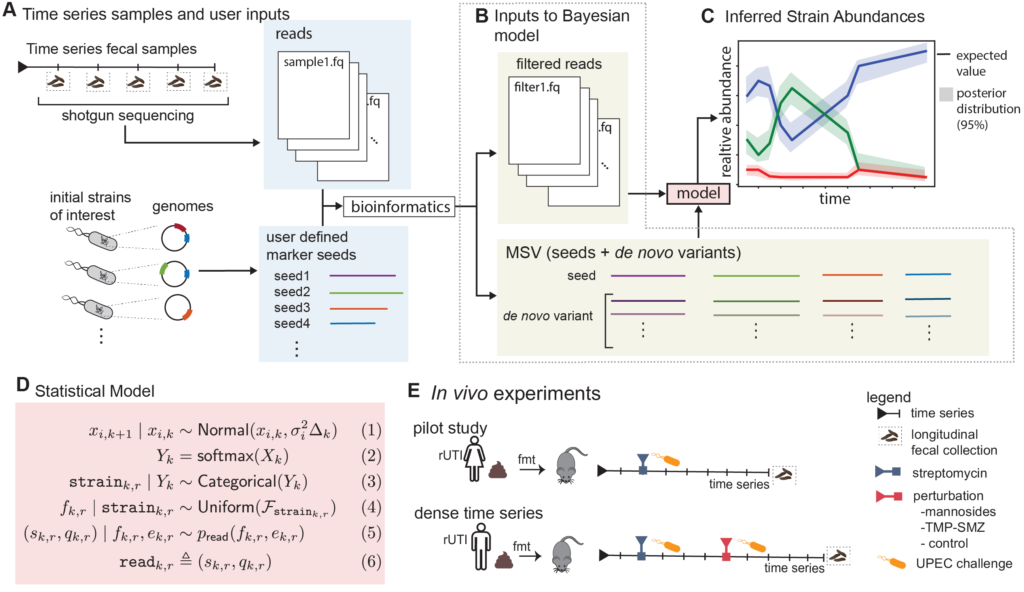
Grant Abstract: Approximately 150 million people annually experience urinary tract infections (UTI), the most common cause of which is uropathogenic Escherichia coli (UPEC). The gut is a known reservoir of UPEC, which typically reside at low abundance, but can transcend the periurethral area to invade the bladder. While the E. coli population within the gut can be diverse, it has been suggested that certain strains have a greater propensity to migrate and cause infection. This may be one driving factor to explain why half of those with an acute infection have a recurrence even after taking antibiotics that clear the first infection from the urinary tract. Being able to detect and track E. coli strains over time would have direct clinical applications for those patients who have frequent recurrences due to gut UPEC carriage. One such clinical application would be early detection and intervention before the onset of infection. Unfortunately, current metagenomic algorithms are not capable of performing strain tracking accurately enough for clinical relevance, especially for low abundance species such as E. coli. A major factor for this lack of accuracy is that all current state-of-the-art metagenomic tools completely ignore temporal dependence between samples. Even if it is known that multiple samples are from the same patient, current tools analyze those samples as if they were independent. Furthermore, many metagenomic tools ignore the sequence quality information that is provided for every nucleobase in every read. We propose to develop a more precise strain tracking algorithm that does take this additional information into account, making the tool host-time-quality aware. Finally, we will pilot and validate our algorithm on a clinically relevant gnotobiotic colonization model. Specifically, humanized germ-free mice will be undergoing two rounds of E. coli challenges with therapeutic perturbations from antibiotics or mannosides, a small molecule precision antibiotic-sparing therapeutic. We propose the following specific aims: (1) Develop the first purpose-built computational method for tracking bacterial strains in the microbiome over time, (2) Gnotobiotic mouse model undergoing UPEC challenges and a therapeutic perturbation. These aims would advance the microbiome field forward allowing for the future development of therapeutics and clinical diagnostics.
Link to NIH Award – Gibson Lab Website